| Search for content and authors |
Density Functional Theory (DFT) study of hydrogen on GaN (0001) surface |
| Pawel Kempisty 1, Pawel Strak 1, Stanisław Krukowski 1,2 |
|
1. Polish Academy of Sciences, Institute of High Pressure Physics (UNIPRESS), Sokolowska 29/37, Warszawa 01-142, Poland |
| Abstract |
| We present ab initio study within the framework of Density Functional Theory (DFT) on the behavior of hydrogen on the GaN (0001) surface depending on the surface coverage and the type of semiconductor doping. We show that the H2 molecules are dissociatively adsorbed on bare GaN(0001) surface with adsorption energy in excess of 2 eV. The adsorption energy is determined by energy change of gallium surface state with respect to energy of bands states at the surface. For bare surface the Fermi level is pinned on surface states equidistant from the top of valence band which is bent at the surface respectively in opposite directions for n-GaN and p-GaN. Therefore, in this case the energy of adsorption does not change significantly and the small differences are due to the effect of the charge localization by different electric field. When surface coverage with hydrogen is about 75% of the monolayer the pinning on surface states disappears. In this situation the type of semiconductor and related Fermi level position is extremely important. We present the dependence of the adsorption energy as a function of hydrogen coverage, which shows a step change in value by about 2 eV when material was changing from n-type to p-type. For the surface coverage close to full monolayer the Fermi level becomes pinned again but in this case at the top of the valence band. Energy shift of the states associated with the adatoms relative to the Fermi level is also independent of the type of doping. Our results are consistent with the results of experiments of Ambacher et al. [1] and Wampler and Myers [2] and help to explain the differences between them.
[1] O. Ambacher et al., Phys. Status Solidi A 159, 105 (1997). 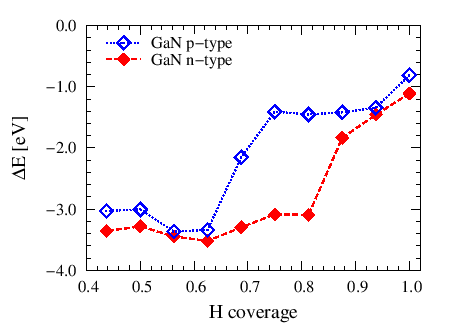 Adsorption energy of single H atom in function of GaN(0001) surface coverage for different type of GaN doping. Adsorption energy of single H atom in function of GaN(0001) surface coverage for different type of GaN doping. |
| Legal notice |
|
| Related papers |
Presentation: Poster at 17th International Conference on Crystal Growth and Epitaxy - ICCGE-17, General Session 1, by Pawel KempistySee On-line Journal of 17th International Conference on Crystal Growth and Epitaxy - ICCGE-17 Submitted: 2013-04-24 16:03 Revised: 2013-04-24 22:52 |