| Search for content and authors |
Problems Associated with Settings for ab initio Calculations of Torsional Barriers with Gaussian 03 |
| Guenter Haefelinger , Frank P. Dietrich |
|
Universität Tübingen, Institut für Organische Chemie, Auf der Morgenstelle 18, Tübingen 72076, Germany |
| Abstract |
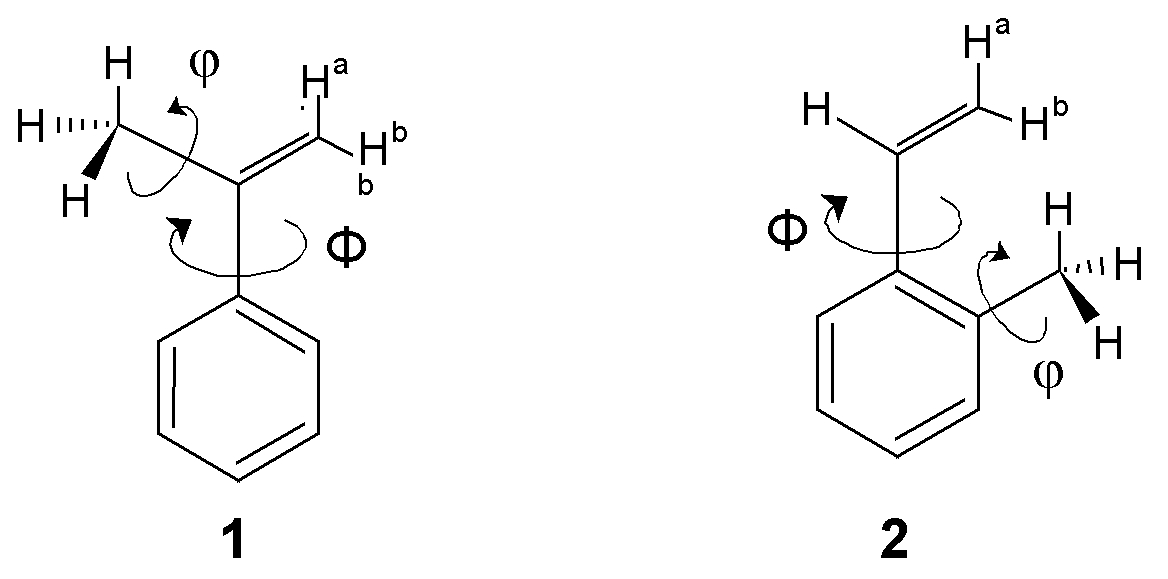
We introduced the use of chemical shifts of geminal protons (Ha and Hb) of vinyl or isopropenyl substituents as experimental and theoretical probe of ring current induced anisotropies of aromatic systems [1].
Difficulties have been observed: 1) for NIMAG-values, 2) with adjustment of extrapolation curves by spline functions for energies and 3) for plots of NMR shieldings while fully optimising the geometry for the complete range of rotation (Φ) between the vinylic substituent and the phenyl system in steps of 15°. The calculation of a two-dimensional energy hypersurface in dependence on the torsional angle Φ and on the rotation of the methyl group φ (with 48 points for 1 and 104 points for 2 in steps of 15° in each dimension) shows how the rotational barrier behaves around the saddle points. The "true" rotational barrier may be extracted as the minimum path on this surface. The observed difficulty can be avoided by optimisations with use of the key words CalcAll (leading to evaluation of the Hessian matrix for each optimisation cycle) or by use of VeryTight (setting extremely tight optimisation convergence criteria). The torsional dependence of experimental and GIAO calculated chemical shifts for the geminal protons Ha and Hb have been compared to predictions by use of our APUDI model [2].
|
| Legal notice |
|
Presentation: poster at 18th Conference on Physical Organic Chemistry, Posters, by Guenter HaefelingerSee On-line Journal of 18th Conference on Physical Organic Chemistry Submitted: 2006-06-06 13:36 Revised: 2009-06-07 00:44 |