| Search for content and authors |
Revealing structural and thermodynamic details of calcium carbonate crystallization intermediates through ab initio methods |
| Raffaella Demichelis , Paolo Raiteri , Julian D. Gale |
|
Nanochemistry Research Institute, Department of Chemistry, Curtin University, PO Box U1987, Perth 6845, Australia |
| Abstract |
Calcium carbonates (CaCO3) play a significant role in the biochemistry of our body and in the geochemistry of our environment, where they are mostly present as a result of biomineralisation. The nucleation and crystal growth processes that lead to the formation of the final crystalline polymorphs have been recently shown to follow pathways alternative to the classical nucleation theory.[1] This has raised the interest in characterising the intermediate phases that appear during the various steps of the process, with the aim of understanding the mechanism itself and the factors driving to the final polymorph differentiation. Computer simulation has the potential of providing useful information in this field, because it allows for an highly accurate exploration of the atomic details and the inter-atomic interactions that are peculiar of a given structure and responsible for its stability. In this respect, ab initio techniques have the advantage of providing the most accurate and transferable models. In fact, they explicitly account for electrons and are independent of empirical parameters, so that once the system is defined all its properties can be calculated straightforward. On the other hand, it is often necessary to find a compromise between accuracy and computing resources, while making sure that the model is realistic. 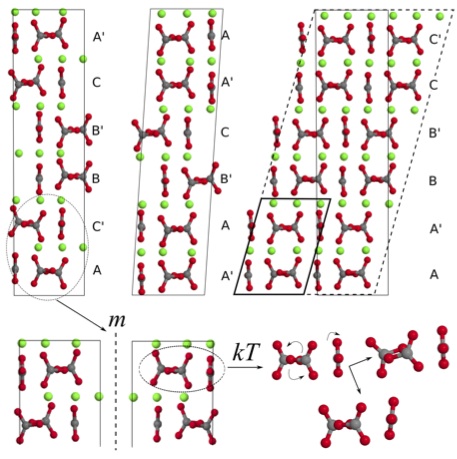 Figure 1: Graphical representation of three isoenergetic structures for vaterite. The different layer stacking sequence, the possible chirality and the rotational freedom of carbonate ions are shown (O, Ca and C atoms are coloured in red, green and grey, respectively). Figure 1: Graphical representation of three isoenergetic structures for vaterite. The different layer stacking sequence, the possible chirality and the rotational freedom of carbonate ions are shown (O, Ca and C atoms are coloured in red, green and grey, respectively).
Density Functional Theory (DFT) is arguably one of the most widely used method for the simulation of crystalline systems. It is implemented in most of the available solid state codes and usually allow to simulate realistic models with a reasonable amount of computing resources. The main disadvantage of DFT is that the exact formulation of the exchange-correlation term in the Hamiltonian is unknown, so that dispersion interactions are not properly accounted for. For this reason, approximations and corrections have to be introduced.[2] Two recent studies that apply DFT to the investigation of the structural and thermodynamic properties of calcium carbonate hydrated and anhydrous phases will be presented, and success and failures will be discussed. The former deals with the major advances that have been achieved through DFT methods in understanding the structure of vaterite.[3] Vaterite is a metastable CaCO3 polymorph whose structure has puzzled scientists for more than half a century. A new model has been suggested, consisting of multiple structures, which establishes a link between the most recent models proposed in the literature.[3,4] The disorder of vaterite is here interpreted in terms of different orientations of the carbonate anions, different stacking sequences of the carbonate layers, and possible chiral forms. Hence, vaterite should be considered as a combination of different forms exhibiting similar average properties, rather than a single “disordered” structure (Figure 1). Furthermore, chirality represents a new and important direction for future investigation that may influence which of the possible vaterite structure is obtained. The latter deals with the relative stability of the CaCO3 anhydrous and hydrated polymorphs as obtained at the DFT level.[5] This study is currently at the limit of DFT capability, because it involves systems exhibiting a very different structure and characterized by different kinds of interactions. In particular, van der Waals interactions are shown to play a non negligible role with respect to ionic and covalent contributions in all of the considered phases. [1] Gebauer et al. (2008) Science, 322, 1819; Meldrum & Sear (2008) Science, 322, 1802; Demichelis et al. (2011) Nat. Commun., 2, 590 [2] Grimme (2004) J. Comput. Chem., 25, 1463; Becke & Johnson (2005) J. Chem. Phys., 122, 154104 [3] Demichelis et al. (2012) CrystEngComm, 14, 44; Demichelis et al. (2013) submitted on February 22nd. [4] Wang & Beker (2009) Am. Mineral., 94, 380; Mugnaioli et al. (2012) Angew. Chem. Int. Ed., 51, 7041 [5] Demichelis et al. (2013) in preparation |
| Legal notice |
|
| Related papers |
Presentation: Oral at 17th International Conference on Crystal Growth and Epitaxy - ICCGE-17, General Session 1, by Raffaella DemichelisSee On-line Journal of 17th International Conference on Crystal Growth and Epitaxy - ICCGE-17 Submitted: 2013-03-21 02:55 Revised: 2013-03-28 04:53 |