| Search for content and authors |
Hydrogen in rutile TiO2 |
| Tor S. Bjørheim , Svein Stølen , Truls E. Norby |
|
University of Oslo, Department of Chemistry, Oslo N-0315, Norway |
| Abstract |
| Rutile TiO2 is a wide-band gap metal oxide semiconductor with a variety of potential applications involving its electrical, optical and catalytic properties. The point defect chemistry governing its properties has been extensively studied since the 1960s, and has recently been reviewed by Nowotny et al.[1]. However, hydrogen defects, such as protons are often neglected. In this study, we utilize ab initio DFT calculations and molecular dynamics (MD) to elaborate on the behaviour of hydrogen defects in rutile TiO2, and predict the relative dominance of protons and oxygen vacancies as positive defects. All calculations were carried out using DFT as implemented in VASP, using GGA-PBE and the PAW method with a cut-off energy of 500 eV. The calculations show that both OHOq and HOq form shallow donor levels (Fig. 1a). Of the three hydrogen defects, protonic defects have the lowest formation energy, even at low oxygen activities, and will thus be the dominating species. However, the formation energy of HO• is only about 0.35 eV higher, and may thus form in small quantities. From the formation energy of OHO• and vO••, we have determined the enthalpy of hydration of oxygen vacancies: H2O(g) + OO× + vO••= 2OHO• to ∆HHydr = -1.57 eV. As such, rutile TiO2 can be expected to be dominated by protons at elevated temperatures, even under relatively dry conditions.
(b)
(c) 1. M.K. Novotny, L.R. Shepard, T. Bak, J. Nowotny, J. Phys. Chem. C 112, (2008), 5275
|
| Legal notice |
|
Presentation: Oral at E-MRS Fall Meeting 2009, Symposium G, by Tor S. BjørheimSee On-line Journal of E-MRS Fall Meeting 2009 Submitted: 2009-05-25 11:53 Revised: 2009-11-25 10:33 |
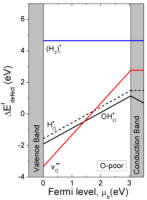 (a)
(a)