| Search for content and authors |
Polymorph prediction by nucleation modeling |
| Rita Bylsma 1, Hugo Meekes 1, Elias Vlieg 2, Joop Horst' ter 3, Niek Klerk' de 1 |
|
1. Radboud university Nijmegen, Solid State Chemistry (RU/SSC), Heyendaalseweg 135, Nijmegen 6525AJ, Netherlands |
| Abstract |
Most organic compounds are polymorphic, i.e. they can crystallize in more than one crystal structure. This explains why crystals of the same compound can exhibit different solubility and effectiveness as a drug. 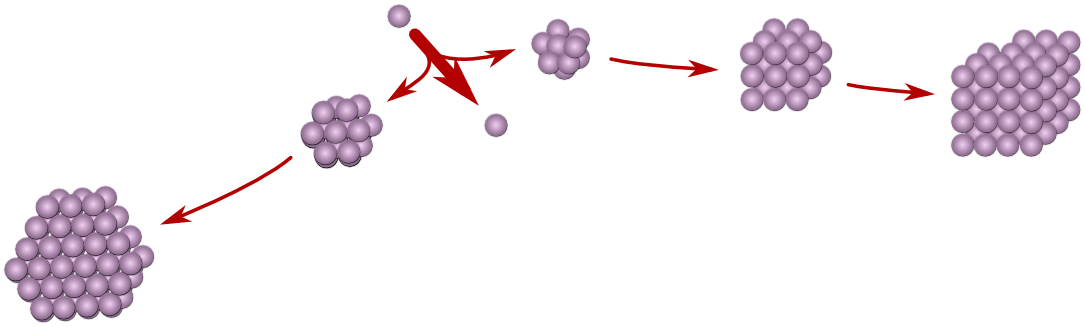
Conventional polymorph prediction routines are based on thermodynamic stability and predict that only one form would exist, the `stable' form. However, due to nucleation kinetics, a thermodynamically unstable form may crystallize and prevent the stable form from crystallizing (Ostwald's rule of stages). Thermodynamics predicts subsequent conversion to the stable form, but as conversion in the crystalline phase is often very slow, unstable polymorphic forms do actually exist over extended periods of time. Therefore, unstable polymorphic forms cannot be ignored in industry and they can even be of use. To be able to predict their formation, we are developing a kinetic polymorph predictor that simulates the nucleation process using a Monte Carlo algorithm.[1] To ensure computability, our kinetic polymorph predictor uses crystal graphs: abstract descriptions of molecular structure, which contain all the important aspects that determine nucleation. We have been using the molecular mechanics program Cerius to compute crystal graphs from crystallographic data. In order to be independent of crystallographic data and to be more accurate, we started using the open source ab initio DFT program Siesta in combination with JuNoLo that adds a vdW-DF-functional. Together they function as a sophisticated energy-based polymorph prediction routine, generating many possible polymorphic structures. We added the functionality of representing them as crystal graphs.
Nucleation would be strongly influenced by the existence of precursor clusters in solution. We therefore added the possibility of simulating nucleation from all possible precursor clusters. We will show results for some polymorphic drugs. |
| Legal notice |
|
| Related papers |
Presentation: Oral at 17th International Conference on Crystal Growth and Epitaxy - ICCGE-17, General Session 1, by Rita BylsmaSee On-line Journal of 17th International Conference on Crystal Growth and Epitaxy - ICCGE-17 Submitted: 2013-04-14 18:24 Revised: 2013-04-15 20:30 |