| Search for content and authors |
HPLC methods for in–process control and chemical purity determination of olopatadine |
| Karolina Kłos , Ewelina Czerniec-Michalik , Joanna Zagrodzka , Katarzyna Badowska-Rosłonek |
|
Pharmaceutical Research Institute (IF), Rydygiera 8, Warszawa 01-793, Poland |
| Abstract |
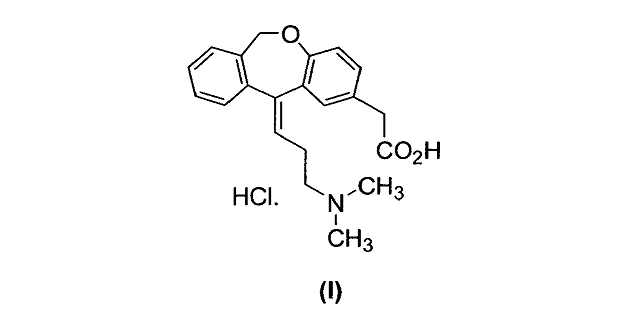
Olopatadine-HCl ([(Z)-3-(Dimethylamino)propylidene]-6-11-dihydrodibenz[b,e] oxepin-2-acetic acid hydrochloride) is a relatively selective H1-receptor antagonist that is used for the treatment of ocular symptoms of seasonal allergic conjunctivitis. The compound represented by formula (I), commonly known as Olopatadine, has been used as an active constituent drug, in form of its hydrochloric salt and may be administered as an ophthalmic solution. During the synthesis of (Z)-olopatadine hydrochloride a number of impurities are formed. The (E) olopatadine isomer is the one of the most significant impurities. The quality control of the starting materials, by-products identification and the purity of active substance are essential in the manufacturing process of drugs. The aim of this control is based on the requirements of ICH and EMA and described in scientific guidelines [1]. Critical points defined during the development are gathered in in-door specifications, taking into account selected parameters important from the synthetic point of view. The main analytical technique used for quantitative determination of impurities in the olopatadine, as well as the in-process control of manufacturing process is HPLC with UV detection. Two HPLC methods were developed to establish purity of drug substance, control synthetic route and the isomerization process of crude final product. The following potential impurities were determined: starting material – 6,11-Di-hydro-11-oxodibenz[b,e]oxepin-2-acetic acid (Isoxepac), p-toluenesulfonic acid, metabolite the (Z)-olopatadine and the (E)-olopatadine isomer. Only one of the compounds mentioned above (isomer E) was considered in the HPLC method recommended by USP [2]. The constant evolution of olopatadine synthesis route demand the adjustment of quality control analytical tools. The HPLC method described in USP is dedicated to samples demonstrating the high degree of purification, published methodology is not suitable for reaction control. Satisfying separation of the API from impurities formed during the production process was achieved on C-8 and C-18 columns using trifluoroacetic acid solution, phosphorous buffer, methanol and acetonitrile as the mobile phases in gradient mode. The flow rate was 1.0 mL min-1 and the detection wavelength was 225 nm. The efficiency of the procedure was verified by its application to standards of potential impurities found during the synthesis development. Presented methods confirmed the acceptance criteria given by Eur. Phar [3]. 1. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Impurities In New Drug Substances Q3A(R2). ICH Secretariat. Geneva. Switzerland (2006) 2. USP Olopatadine Hydrochloride, USP34, p. 3711 3. European Directorate for the Quality of Medicines & HealthCare. European Pharmacopoeia 7.0. Chapter: 2.2.46 Chromatographic separation techniques (2010) |
| Legal notice |
|
| Related papers |
Presentation: Poster at VIII Multidyscyplinarna Konferencja Nauki o Leku, by Ewelina Czerniec-MichalikSee On-line Journal of VIII Multidyscyplinarna Konferencja Nauki o Leku Submitted: 2012-03-26 14:27 Revised: 2012-05-15 18:46 |